大数据文摘授权转载自AI科技评论
作者 | 李梅、黄楠
编辑 | 陈彩娴
2018 年,Deepmind 首次发布基于深度神经网络的蛋白质结构预测数据库 AlphaFold,在蛋白质预测中实现了最先进的性能;去年,AlphaFold 2 获得了 98.5% 的蛋白质预测率;前段时间,Deepmind 又重磅发布了数据集更新,称目前的 AlphaFold 已经预测了几乎所有已知的蛋白质。
如何有效识别****物作用机制在今天仍然是一个巨大挑战,计算对接的方法已被广泛用于预测****物结合靶点。有了大规模蛋白质结构预测技术,****物发现将变得更容易。所以,自 AlphaFold 问世以来,称其将引发一场结构生物学的革命、彻底改变****物发现的声音就不绝于耳。
本质上,AlphaFold 是一个工具,我们目前真的能利用好这个工具吗?
近日,来自 MIT 的研究团队给出了否定的回答。
他们对使用 AlphaFold2 的分子对接模拟的模型性能进行了评估,发现模型在识别真正的蛋白质-配体相互作用方面的预测能力较弱,并证明需要使用基于机器学习的方法进行建模来提高模型性能,以更好地利用AlphaFold2 进行****物发现。该论文“Benchmarking AlphaFold-enabled molecular docking predictions for antibiotic discovery”发表在了Molecular Systems Biology 期刊上。

论文地址:https://www.embopress.org/doi/epdf/10.15252/msb.202211081
使用AlphaFold 2 预测分子对接
所谓化合物的对接计算,是将候选化合物列表中的每一个对接到目标蛋白质中,生成最有可能结合的化合物的粗略排序。这个过程可以在化合物集合上完成,获得庞大的虚拟库,这种虚拟筛选已经成为计算化学领域的长期目标。筛选 218 种大肠杆菌活性化合物研究团队首先筛选了一组化合物,包含大约 39128 种,其中包括已知****物(已知抗生素)、活性天然产物和一系列其他不同结构,并在针对大肠杆菌的筛选中发现了 218 种化合物培养物。仅仅有 218 个阳性,这个结果是令人惊讶的,但考虑到抗菌****物发现工作的难度,这个数字也算比较难得了。在 218 种活性化合物中,有大约 80% 是已知抗生素类别中的成员,剩下的部分则是已知细胞毒性化合物和一些新的通配类型的混合。这为实验的进行提供了一个很好的背景,因为在大多数情况下,我们可以预测从反向对接筛选中获得什么结果。将活性化合物与预测的蛋白质结构对接接着,团队研究了这些活性化合物的潜在结合靶标。多年来,在大肠杆菌中进行的大量基因组敲除扫描的共识评分已经得出了 296 项基本蛋白质,所以,可以合理推断所有真正抑制生长的靶标蛋白质都可能在这些列表当中。作者将 218 种活性化合物中与 AlphaFold 2 预测出的 296 种基本大肠杆菌蛋白质结构进行对接,并通过几种不同的计算方法,对 218 种化合物与 296 种蛋白质的组合进行计算,预测了 64000 多个蛋白质-配体对的结合位姿与结合亲和力预测。 图注:在 AlphaFold 结构上进行分子对接从计算量来看,这是一个可靠度相当高的测试,尤其是考虑到内部控制的数量(具有已知靶标的化合物和在这些靶标内具有已知结合构象的化合物),这项计算是很有价值的。作为对照,研究团队还从一组化合物中,随机选择了 100 种对细菌生长完全没有抑制作用的化合物进行相同的计算,从而获得对 29600 个蛋白质-配体对的结合位姿与亲和力预测。
图注:在 AlphaFold 结构上进行分子对接从计算量来看,这是一个可靠度相当高的测试,尤其是考虑到内部控制的数量(具有已知靶标的化合物和在这些靶标内具有已知结合构象的化合物),这项计算是很有价值的。作为对照,研究团队还从一组化合物中,随机选择了 100 种对细菌生长完全没有抑制作用的化合物进行相同的计算,从而获得对 29600 个蛋白质-配体对的结合位姿与亲和力预测。基于 AlphaFold 2 预测结构的模型性能很弱
虽然这项工作预测了包括活性和非活性化合物的化合物与蛋白质混杂性,但问题是,这些预测中有多少是假阳性?将模型预测与已知的抗生素结合目标进行比较为了评估所用模型方法的性能,作者将模型预测与常用抗生素类别的已知相互作用进行比较。作者搜集了先前文献中的抗生素-蛋白质靶对,组成一个包含 142 种抗生素-蛋白质相互作用的数据集。结果发现,他们的模型仅仅正确预测了 3 种具有强结合性(即结合亲和力阈值为 -7 kcal/mol )的相互作用,以及 43 种具有一般结合性(即结合亲和力阈值为 -5 kcal/mol )的相互作用。所以,模型预测的真阳性率分别为 2.1% 和 30.3%。这种比较表明,基于 AlphaFold 2 预测结构的建模平台性能很弱。测量 12 种基本蛋白质的酶抑制作者接着选取了 12 种基本蛋白质,它们可以用于酶促测定,通过测量 218 种活性化合物对这些蛋白质的酶抑制,作者对模型预测的子集进行进一步的评估。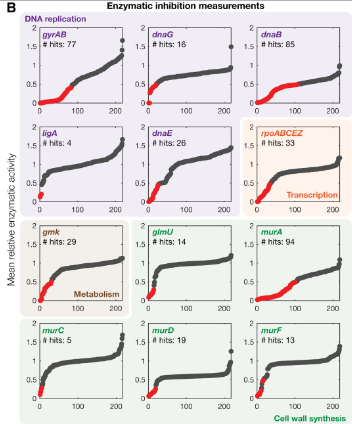 图注:所有 218 种活性化合物的平均相对活性,12 种蛋白质都经过抑制实验测试。结合相互作用命中是蛋白质-配体相互作用(红点),所有其他交互都被指定为非命中(灰点)。结果表明,所有测试中的基本蛋白质都被至少四种不同的化合物所抑制,涵盖从强到弱的一系列结合亲和力阈值,基于 AlphaFold 2 的模型所预测的对接显示出了广泛的混杂性。模型的基准测试最后,作者对建模平台的性能进行了统计基准测试。基于酶抑制测量数据,作者将实验观察到的结合相互作用命中与他们预测的相互作用进行比较,结合亲和力阈值设定为小于 -5 kcal/mol 和 -7 kcal/mol。结果发现,更强结合性的亲和力阈值会导致更少的结合相互作用预测,此时真阳性率更低,准确性更高。模型性能根据所设定的结合亲和力阈值从弱到中等变化。作者又在独立于结合亲和力阈值的条件下,使用接受者操作特征曲线 (ROC) 和精确召回曲线 (PR) 再次进行评估,也都表明模型性能较弱。总之,模型的预测结果中不仅存在大量假阳性(即非活性化合物被预测为与关键细菌蛋白质的活性位点结合),而且还有大量的假阴性(即已知存在相互作用,但没有被发现)。只有达到最严格的结合亲和力阈值时,模型才会比随机预测表现得略好一些。
图注:所有 218 种活性化合物的平均相对活性,12 种蛋白质都经过抑制实验测试。结合相互作用命中是蛋白质-配体相互作用(红点),所有其他交互都被指定为非命中(灰点)。结果表明,所有测试中的基本蛋白质都被至少四种不同的化合物所抑制,涵盖从强到弱的一系列结合亲和力阈值,基于 AlphaFold 2 的模型所预测的对接显示出了广泛的混杂性。模型的基准测试最后,作者对建模平台的性能进行了统计基准测试。基于酶抑制测量数据,作者将实验观察到的结合相互作用命中与他们预测的相互作用进行比较,结合亲和力阈值设定为小于 -5 kcal/mol 和 -7 kcal/mol。结果发现,更强结合性的亲和力阈值会导致更少的结合相互作用预测,此时真阳性率更低,准确性更高。模型性能根据所设定的结合亲和力阈值从弱到中等变化。作者又在独立于结合亲和力阈值的条件下,使用接受者操作特征曲线 (ROC) 和精确召回曲线 (PR) 再次进行评估,也都表明模型性能较弱。总之,模型的预测结果中不仅存在大量假阳性(即非活性化合物被预测为与关键细菌蛋白质的活性位点结合),而且还有大量的假阴性(即已知存在相互作用,但没有被发现)。只有达到最严格的结合亲和力阈值时,模型才会比随机预测表现得略好一些。AlphaFold 本身没错,用好机器学习方法是关键
接下来的问题是,模型的弱性能是由 AlphaFold2 所提供的蛋白质结构质量导致的吗?问题出自对接方法而非蛋白质结构质量为了验证这个问题,作者将 218 种活性化合物与八种实验确定的蛋白质结构中的每一种对接进行了重复的对接模拟,并同样对模型性能进行了基准测试,结果是 auROC 值在数量上与先前相似,范围从 0.25 ( glmU ) 到 0.69 ( gyrAB ),平均值为 0.46。auPRC 值也发现了类似的结果,范围从 0.03 ( ligA ) 到 0.56 ( gyrAB ),平均值为 0.22。这些发现表明,使用 AlphaFold2 预测结构的分子对接与使用实验确定的结构是类似的。这也与之前对 AlphaFold 对实验确定的蛋白质结构的保真度评估一致,由此可以得出,模型的性能弱是因为对接方法的原因,而不是蛋白质结构的质量差。使用机器学习方法可改进模型性能基于分子对接的弱性能问题,研究团队探索了可以提高性能的方法。研究中使用了四种不同的基于机器学习的评分函数,分别是 RF-Score 、RF-Score-VS、PLEC score 和 NNScore,以对模型性能进行基准测试和改进。相比于 RF-Score 和 RF-Score-VS - RF-Score 的虚拟筛选适应性--利用随机森林或决策树的组合来预测蛋白质与配体的结合亲和力,PLEC score 采用了蛋白质-配体对之间的扩展连接指纹,NNScore 是基于神经网络的集合。作者在研究中采用了评分函数,使用 PDBbind v2016 或有用的诱饵目录对增强(DUD-E)数据库进行训练,以重新评估 AutoDock Vina 预测的对接姿势。此外,研究使用 DOCK6.9 和应用于 AutoDock Vina 姿势的每个基于机器学习的评分函数,还预测了每种抗菌化合物与 12 种经验测试必需蛋白中每一种之间的结合亲和力,并对每种方法的性能进行基准测试。测试结果发现,平均 auROC 值在 0.46 和 0.63 之间(下图 A)。其中,与 DOCK6.9 对接并使用 PLEC score 对 AutoDock Vina 姿势进行重新评分平均,导致 auROC 值低于单独使用 AutoDock Vina 的结果,DOCK6.9 的 auROC 值为为 0.46(范围为 0.25 至 0.61)和 0.47(范围 PLEC score 为 0.28 至 0.63)(下图 A)相比之下,使用 RF-Score、RF-Score-VS 或 NNScore 对 AutoDock Vina 姿势进行重新评分可提高模型性能,平均 auROC 值分别为 0.62(范围为 0.53 至 0.69)、0.63(范围为 0.46 至 0.75)和 0.58(范围为 0.41 到 0.69)。研究结果也与 auPRC 相似,当使用 RF-Score 重新评分时,其平均值高达 0.24。这些模型性能评估表明,某些基于机器学习的评分函数提高了预测准确性。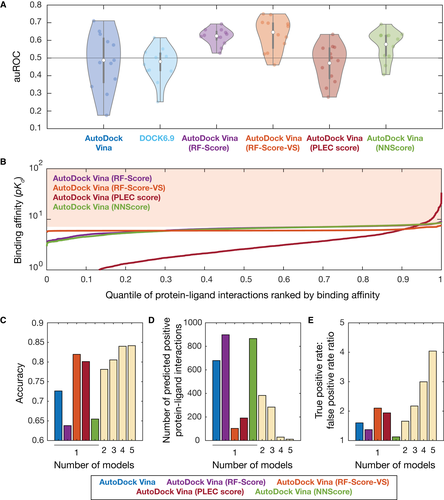 图注:使用机器学习对模型性能进行基准测试和改进。A. 在不同的分子对接程序和不同的基于机器学习的姿势评分函数。白点表示平均值;灰色条表第25-75个百分位值的范围;灰色箱线图须线表示不被视为异常值的值范围;0.5 处的水平线表示随机预测生成的基准。B. 通过在 AutoDock Vina 应用基于机器学习的重新评分函数建模的蛋白质-配体对的排序结合亲和力 。曲线根据 (A) 中使用的重新评分函数着色;阴影区域表示 > 7 的结合亲和力阈值。C-E. 预测准确性、预测阳性数(蛋白质-配体相互作用)和真阳性率/假阳性率对所用模型数量的依赖性。群体智慧方法可提高预测准确性由于某些基于机器学习的评分函数会增加 auROC 和 auPRC,研究还探讨了在严格限制结合亲和力阈值的情况下,结合“群体智慧”方法使用重新评分模型,是否可以提高预测准确性和真阳性率。作者将预测的蛋白质-配体相互作用,定义为满足所有模型的结合亲和力阈值,并将 AutoDock Vina 预测与上述四种基于机器学习的评分函数的预测相结合;研究通过使用这种共识方法发现,预测准确性可随着使用的模型数量而提高(上图 C),这同预测的蛋白质-配体相互作用数量的相应减少预期一致(上图 D)。与此同时,真阳性率与假阳性率的比率则是随使用模型数量的增加而增加,在预期之外(上图 E)。可以看到,该结果同使用某些基于机器学习的评分函数提高预测能力的发现一致,这也进一步表明了,将分子对接与基于机器学习的模型结合起来,可以让人们更好地利用 AlphaFold2 预测的蛋白质结构进行****物筛选。所以,一些机器学习方法确实可以提高了预测的准确性。不过,这只是部分的成功,当前研究所用的数据集中有很多已经确定的蛋白质和化合物的实验事实,如果涉及那些较少被关注的领域,这些方法是否仍然奏效就不可知了。虽然 AlphaFold 为我们提供了大量且合理的蛋白质结构,但我们实现它的价值的能力还非常有限。所以至少在目前看来,“AlphaFold 将彻底改变****物发现”的说法还尚待证实,成功还在未来。参考链接:https://www.science.org/content/blog-post/not-alphafold-s-fault
图注:使用机器学习对模型性能进行基准测试和改进。A. 在不同的分子对接程序和不同的基于机器学习的姿势评分函数。白点表示平均值;灰色条表第25-75个百分位值的范围;灰色箱线图须线表示不被视为异常值的值范围;0.5 处的水平线表示随机预测生成的基准。B. 通过在 AutoDock Vina 应用基于机器学习的重新评分函数建模的蛋白质-配体对的排序结合亲和力 。曲线根据 (A) 中使用的重新评分函数着色;阴影区域表示 > 7 的结合亲和力阈值。C-E. 预测准确性、预测阳性数(蛋白质-配体相互作用)和真阳性率/假阳性率对所用模型数量的依赖性。群体智慧方法可提高预测准确性由于某些基于机器学习的评分函数会增加 auROC 和 auPRC,研究还探讨了在严格限制结合亲和力阈值的情况下,结合“群体智慧”方法使用重新评分模型,是否可以提高预测准确性和真阳性率。作者将预测的蛋白质-配体相互作用,定义为满足所有模型的结合亲和力阈值,并将 AutoDock Vina 预测与上述四种基于机器学习的评分函数的预测相结合;研究通过使用这种共识方法发现,预测准确性可随着使用的模型数量而提高(上图 C),这同预测的蛋白质-配体相互作用数量的相应减少预期一致(上图 D)。与此同时,真阳性率与假阳性率的比率则是随使用模型数量的增加而增加,在预期之外(上图 E)。可以看到,该结果同使用某些基于机器学习的评分函数提高预测能力的发现一致,这也进一步表明了,将分子对接与基于机器学习的模型结合起来,可以让人们更好地利用 AlphaFold2 预测的蛋白质结构进行****物筛选。所以,一些机器学习方法确实可以提高了预测的准确性。不过,这只是部分的成功,当前研究所用的数据集中有很多已经确定的蛋白质和化合物的实验事实,如果涉及那些较少被关注的领域,这些方法是否仍然奏效就不可知了。虽然 AlphaFold 为我们提供了大量且合理的蛋白质结构,但我们实现它的价值的能力还非常有限。所以至少在目前看来,“AlphaFold 将彻底改变****物发现”的说法还尚待证实,成功还在未来。参考链接:https://www.science.org/content/blog-post/not-alphafold-s-fault
*博客内容为网友个人发布,仅代表博主个人观点,如有侵权请联系工作人员删除。


